中心思想
本报告基于PharnexCloud网站提供的资料,分析了中国化学药品注册上市的临床要求。核心观点如下:
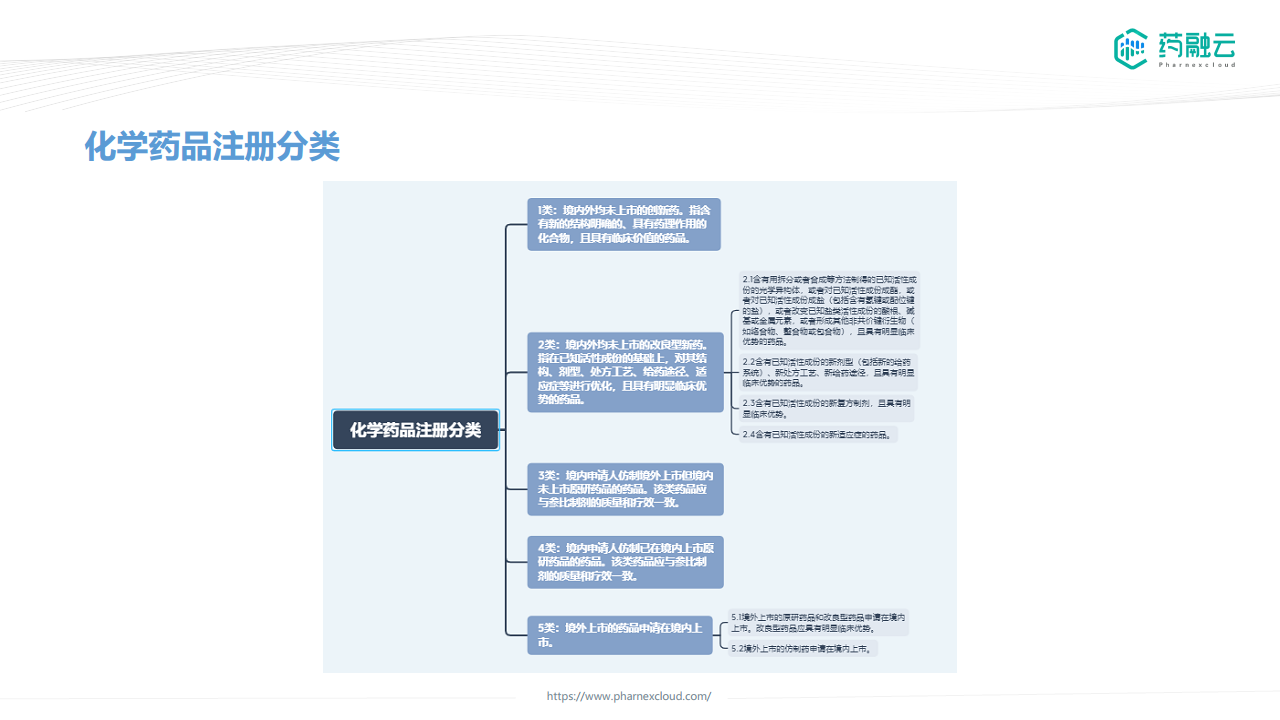
- 中国化学药品注册分类分为1类创新药、2类改良型新药、3类仿制药和4类仿制药,以及5类境外药品,其临床研究要求差异显著。
- 不同类型的化学药品,其临床研究阶段、所需研究类型(药代动力学、药效学、生物等效性研究等)、以及所需临床试验规模和设计均存在较大差异。
- 对于仿制药,尤其4类仿制药,根据剂型(口服、注射、外用等)的不同,临床研究要求也存在显著差异,部分剂型可豁免生物等效性研究。
- 对于特殊类型药品(如临床急需药品、儿童用药、罕见病用药),国家药监局提供优先审评和附条件上市的政策支持。
- 部分指导原则仍处于征求意见稿或草案阶段,药品注册申报过程中,建议与国家药品监督管理局(CDE)进行充分沟通。
主要内容
不同类型化学药品的临床研究要求
本报告根据药品注册分类,详细阐述了不同类型化学药品的临床研究要求:
1类创新药
1类创新药的临床研究遵循四个阶段(I-IV期)的经典模式。I期主要关注安全性、耐受性、药代动力学和药效学;II期和III期进行探索性和确证性研究,评估安全性与有效性,并开展特殊人群研究;IV期为上市后研究。 相关指导原则包括《关于公开征求ICH《E8(R1):临床研究的一般考虑》》、《化学药创新药临床单次和多次给药剂量递增药代动力学研究技术指导原则》等多个文件。
2类改良型新药
2类改良型新药需要体现临床优势,通常需要进行临床前和临床研究。由于产品类型复杂,CDE提倡针对具体品种进行临床问题的沟通。相关指导原则包括《化学药品改良型新药临床试验技术指导原则》和《改良型新药调释制剂临床药代动力学研究技术指导原则》。
3类仿制药
目前缺乏明确的3类仿制药申报指南。参照《境外已上市境内未上市药品临床技术要求》,通常需要开展必要的临床试验以支持仿制药在中国患者中的安全性和有效性评价。3类首仿可能需要进行与原研药物的等效性研究以及额外的临床研究。申报前建议与CDE沟通。
4类仿制药
4类仿制药的临床要求根据剂型而异:
- 口服制剂: 通常进行空腹和餐后生物等效性研究,部分情况可豁免。 相关指导原则包括《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《调释制剂仿制药生物等效性研究要求》等。
- 注射剂: 普通注射剂(真溶液)无需临床研究,特殊注射剂(如脂质体、微球等)则可能需要非临床研究、BE研究,甚至随机对照临床试验。相关指导原则包括《化学药品注射剂仿制药质量和疗效一致性评价技术要求》等。
- 皮肤外用化学仿制药: 需要非临床研究(皮肤刺激性试验、皮肤过敏试验等),必要时进行局部药代动力学和系统暴露量比较研究。部分情况可豁免BE研究。相关指导原则包括《皮肤外用化学仿制药研究技术指导原则》。
- 局部给药局部起效药物: 可根据情况开展必要的临床对比研究,包括PK/PD生物等效性研究和临床指标等效性研究。相关指导原则包括《局部给药局部起效药物临床试验技术指导原则》。
- 经口吸入制剂: 吸入溶液剂可能无需人体生物等效性研究,吸入混悬剂、气雾剂、粉雾剂则通常需要进行人体生物等效性研究,甚至随机对照临床试验。相关指导原则包括《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《境外已上市境内未上市经口吸入制剂仿制药临床试验技术指导原则》等。
5类药物
5类药物主要指境外药品:
- 5.1类境外原研药品: 鼓励在中国同步开展临床试验。根据种族敏感性情况,可能需要开展桥接性临床试验。
- 5.2类境内外仿制药品: 基于原研药品临床评价结果,开展必要的中国患者人群临床试验。对于经口吸入制剂,需要进行生物等效性研究,并根据获益风险特征评估是否需要额外临床试验。相关指导原则包括《境外已上市境内未上市药品临床技术要求》等。
特殊类型药品的临床研究要求
对于临床急需药品、儿童用药和罕见病用药,国家药监局提供优先审评和附条件上市的政策支持。相关指导原则包括《临床急需药品附条件批准上市技术指导原则》、《儿童用化学药品改良型新药临床试验技术指导原则(试行)》等。
总结
中国化学药品注册上市的临床要求复杂且多样化,不同类型的药品,其临床研究要求存在显著差异。 本报告总结了不同类型化学药品的临床研究要求,并特别强调了4类仿制药根据不同剂型的具体要求,以及特殊类型药品的优先审评政策。 由于相关指导原则仍在不断更新和完善,建议药品研发企业在申报前仔细研读相关法规,并与CDE进行充分沟通,以确保注册申报的顺利进行。 未来,随着更多指导原则的出台和完善,以及监管经验的积累,中国化学药品的注册上市流程有望更加规范和高效。



 微信扫一扫-立即使用
微信扫一扫-立即使用